ATAC-seq
背景
染色质开放性 Chromatin Accessibility
人的DNA链全部展开大约有2m,需要折叠为染色质结构才可以存储到放到细胞核中。染色质的基本结构单位是核小体,核小体再折叠能形成高度压缩的染色质结构。这个过程像我们将文件压缩为zip或者rar的压缩包,只要在使用的时候才会解压出来,平时可以减少它的占用空间。
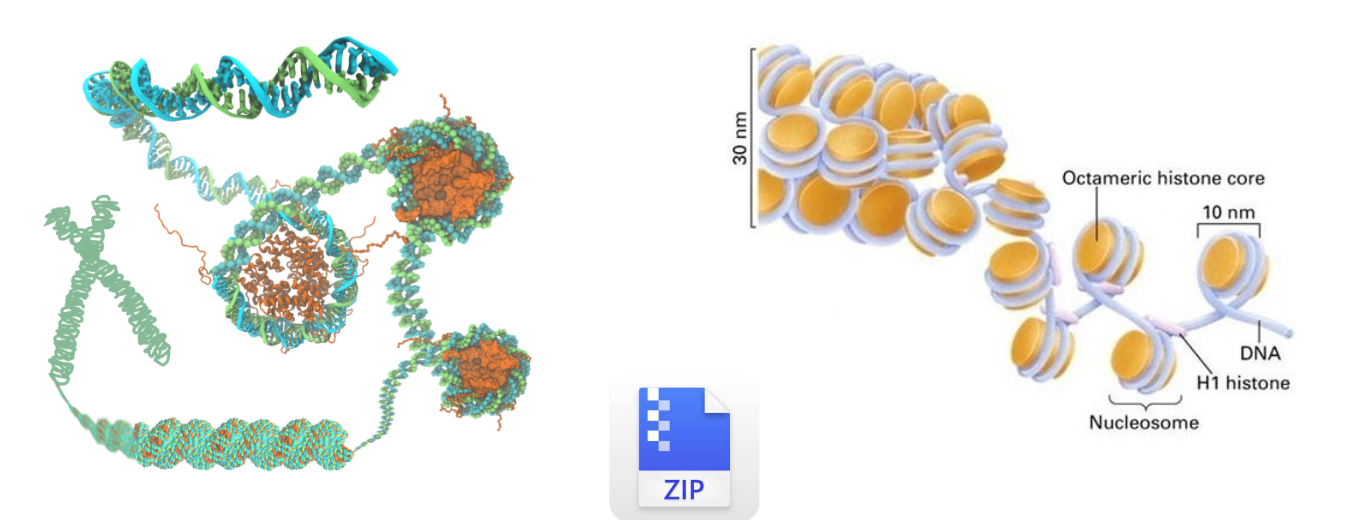
高度折叠的染色质结构在复制和转录时需要暴露出DNA序列,这段暴露的区域就是染色质开放区域,这个区域可以供转录因子和其他调控元件结合,所以它与转录调控是密切相关的。
因此这种致密的核小体结构被破坏后,启动子、增强子等顺式调控元件和反式作用因子可以接近的特性,叫染色质开放性(Chromatin Accessibility)。
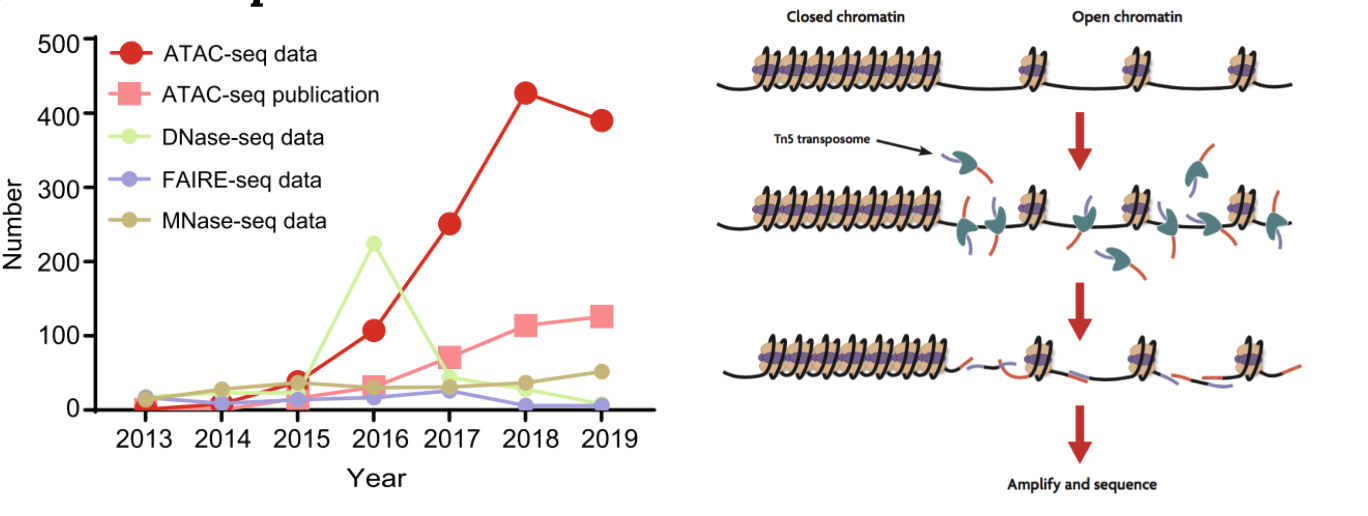
为了研究染色质的开放性,目前有MNase-seq,Dnase-seq,ATAC-seq等,但是目前最常用的是2013年由斯坦福大学开发的ATAC-seq。与传统的MNase-seq以及DNase-seq相比,其具有可重复性强,实验步骤简单,需要的实验样本量少等优点,因而被广泛应用1。
原理
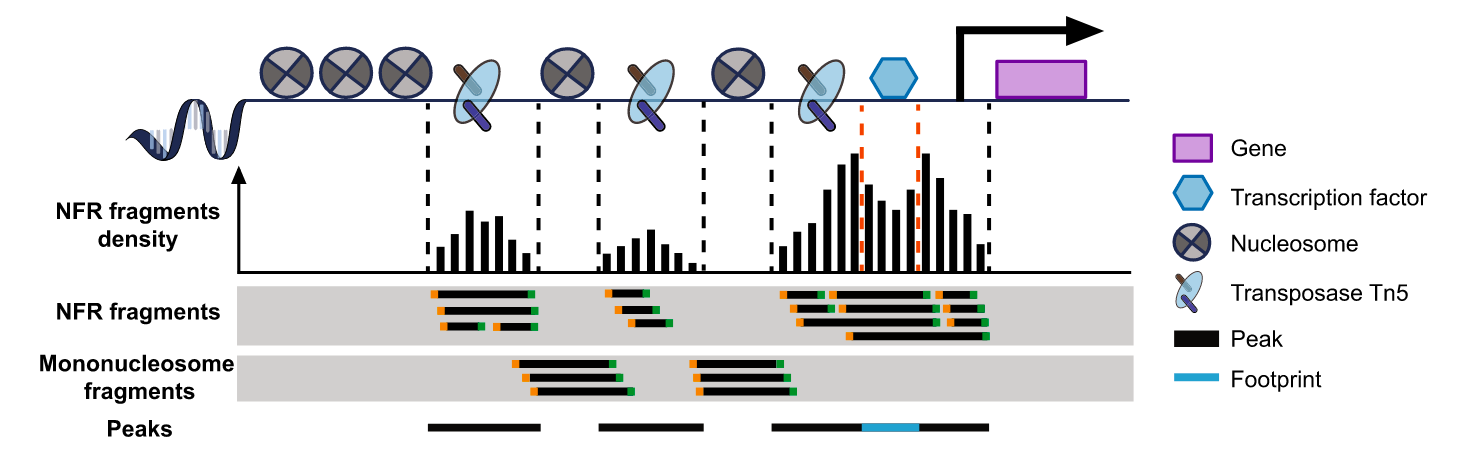
利用转座酶Tn5会携带特定的已知序列,并且可以结合开放的染色质。Tn5酶对染色质开放区进行打断,在打断的同时加上测序接头,接着进行DNA提取,PCR扩增构建文库。经过测序分析,就可以推断染色质可行性、转录因子结合位点、组蛋白修饰区域和核小体位置。
研究内容
肠上皮化生简称肠化,是指正常的胃黏膜上皮被肠型上皮所取代。
正常情况下,我们的器官各司其职,胃表面生长的是具有分泌胃酸功能的胃黏膜上皮细胞,肠道表面生长的是具有分泌和吸收功能的肠黏膜上皮细胞。但当胃黏膜细胞受到比较严重的损伤后,胃肠黏膜上皮结构出现了一定改变,越长越像邻居家肠黏膜的孩子。看上去就像肠黏膜长错了地方,本该长在肠道上长的结构却出现在了胃黏膜上,就像一片草地长出了树木,树木就显得很突出。

目前的假设是,胃黏膜腺体的颈部干细胞具有多方面分泌的潜能,在正常时它可以分化成各种胃黏膜的成熟上皮细胞[9]。干细胞不正常工作时肠化进程会加速,从肠化生过渡到胃癌,而肠化属于胃癌前病变的一种。
胃黏膜上皮细胞癌变并非一朝一夕的事情,不是由正常细胞一跃成为癌细胞,而是一个慢性渐进的过程,在发展成恶性肿瘤之前,经历多年持续的癌前变化。若能及早识别和及早干预,也是一种防止胃癌的有效途径。
因此从干细胞水平上能够发现促使正常干细胞分化为肠化细胞的根本原因,对于预防胃癌,以及使肠化逆转显得尤为重要。
实验设计:
针对10个病人,分别采集胃,肠化组织,分为两组(stemness + / - )进行培养。
其中阴性对照:正常胃组织,阳性对照:正常十二指肠组织,
stemness: 位置细胞干性的条件。+ 维持干性,-不维持,IM: + 。 - 。
胃窦:A, 胃体:C, 胃角:AC
分析
目前已经开源了很多ATAC-seq原始data的预处理与计算,其基本流程为:
QC->Alignment->Remove low quality-> Call Peak
针对Call Peak 的结果,可以计算不同组间差异的Peak,或者Motif 富集与转录因子足迹分析,更进一步的可以联合RNA-seq。
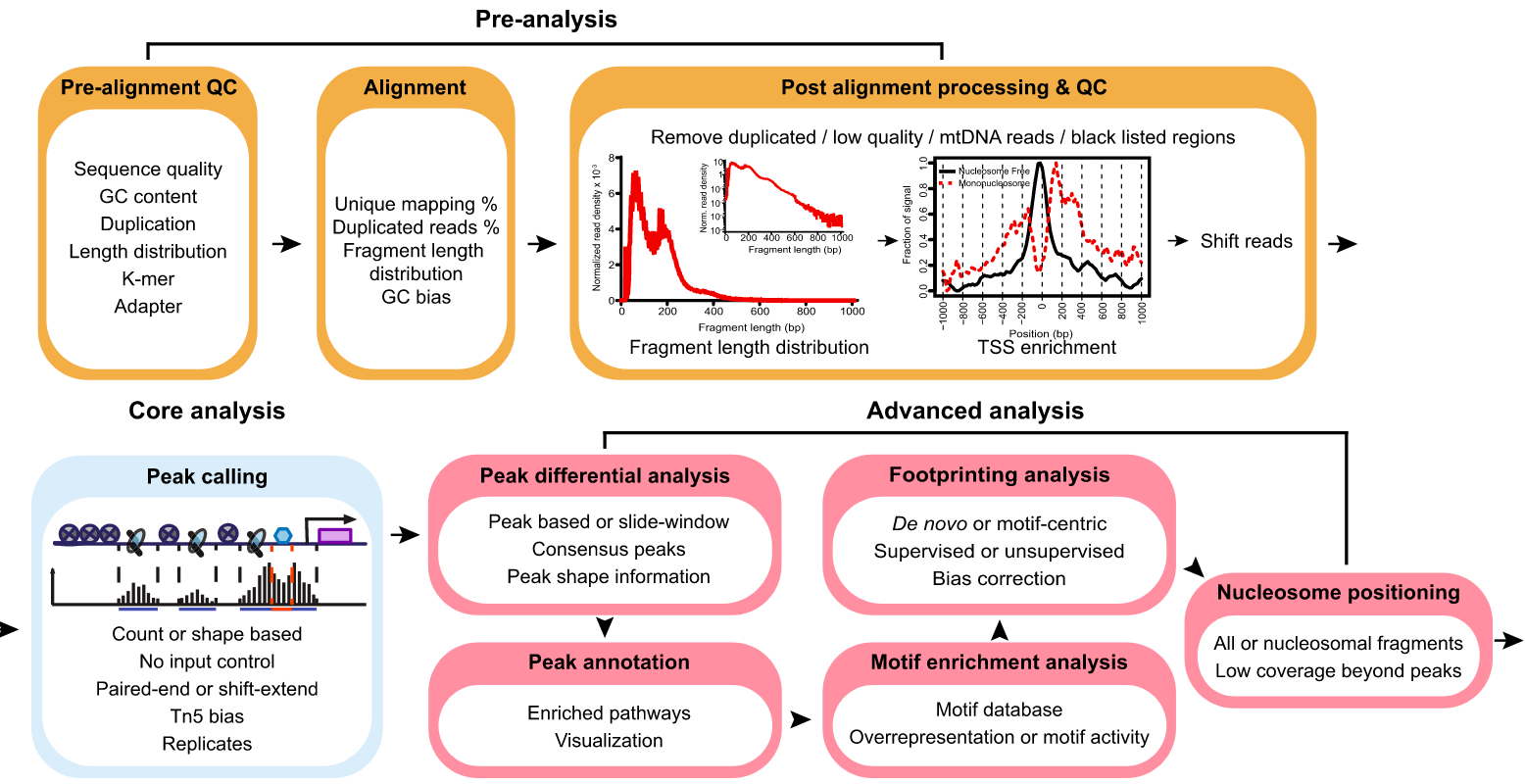
Pipeline:
This pipeline is designed for automated end-to-end quality control and processing of ATAC-seq and DNase-seq data.
标准
介绍前期质控指标,避免样本问题对后期实验结果的影响,造成错误或返工
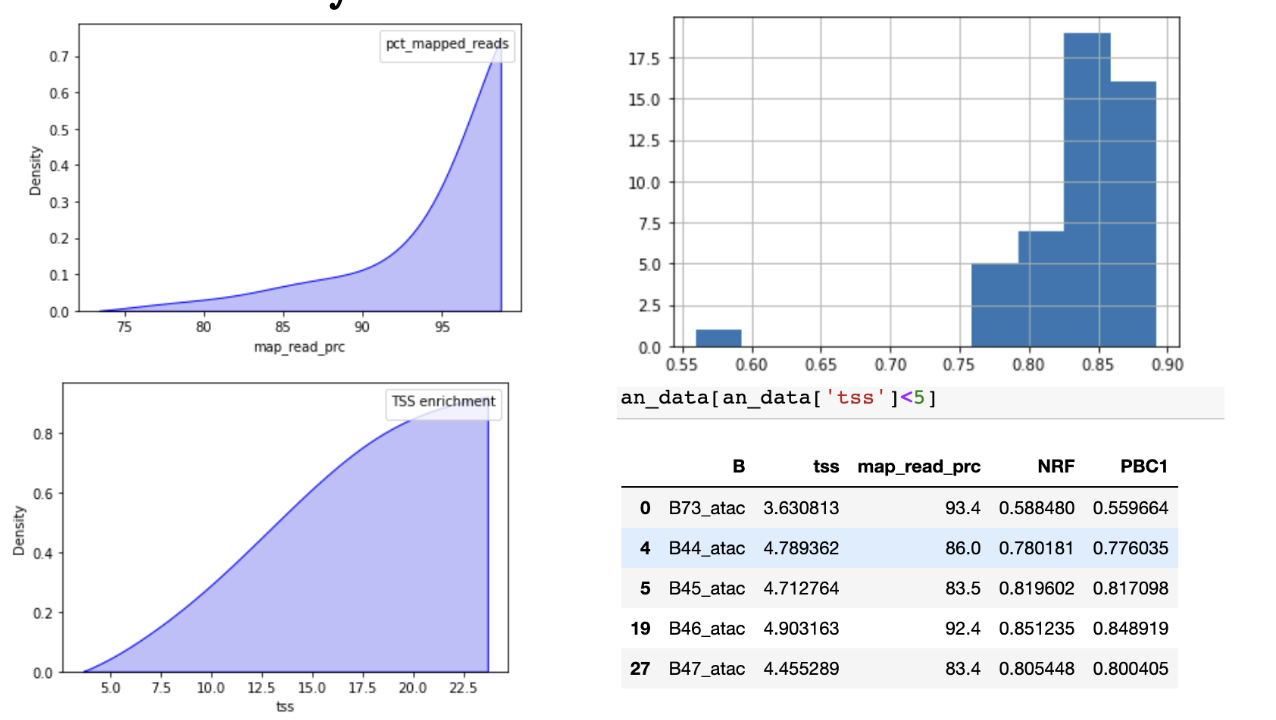
比对率:
正常是超过95%,最低不能低于80%。
1 | 可以抽取部分样本在nt数据库中进行比对,看map到那些物种中,是否有部分细菌污染 |
峰值区域的读数比例(FRiP score):
FRiP score应大于0.3,最低不能低于0.2。
所有映射的读数中,属于被称为峰值区域的部分,即显著富集的峰值中的可用读数除以所有可用读数。一般来说,FRiP得分与区域的数量呈正相关.(Landt et al, Genome Research Sept. 2012, 22(9): 1813–1831)
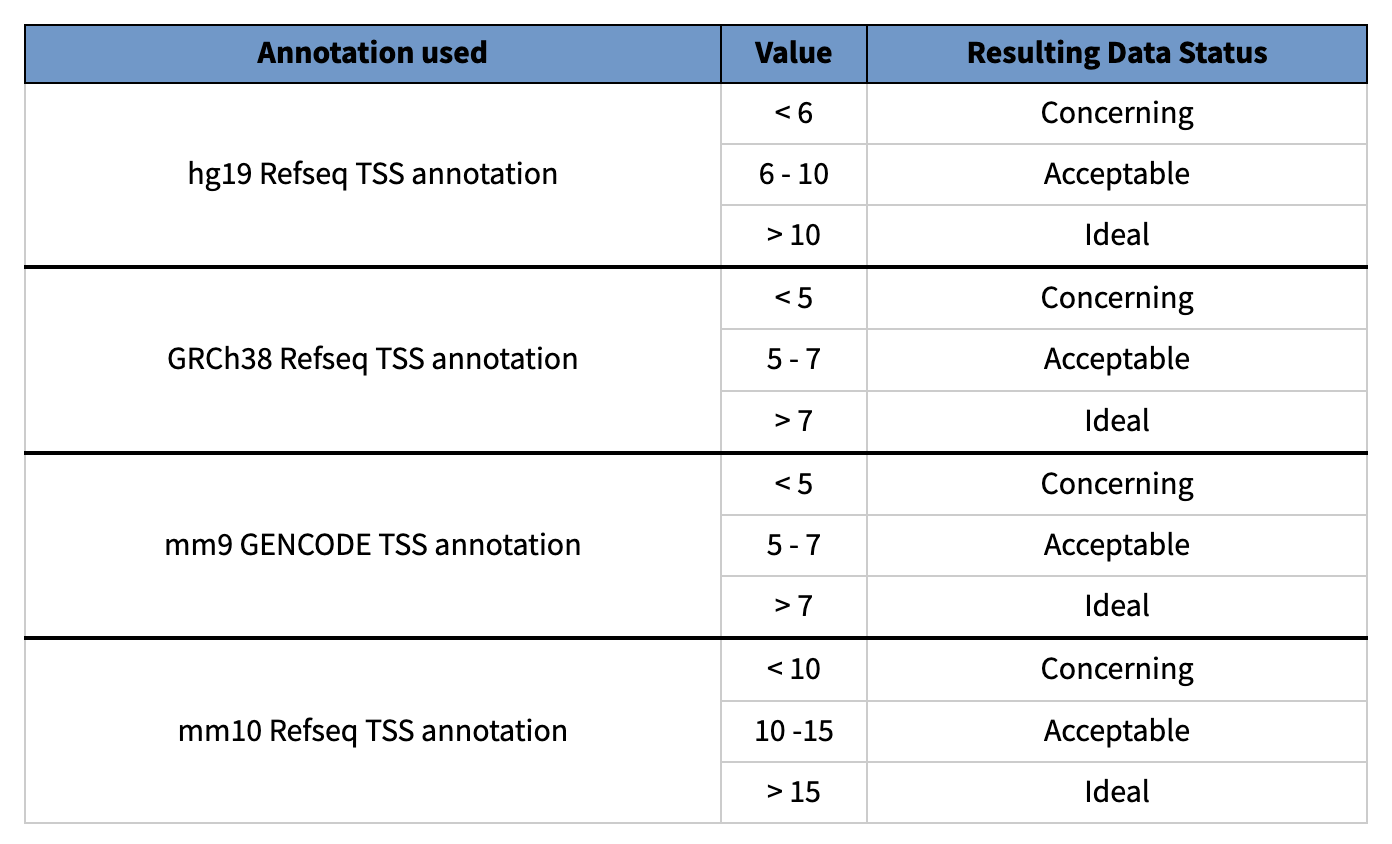
TSS 富集:
TSS富集计算是一种信噪比计算。收集一组参考TSSs周围的读数,形成以TSSs为中心、向任一方向延伸2000bp(共计4000bp)的读数总分布。然后,该分布被归一化,即在分布的每个末端侧翼的100bps内取平均读数深度(总共200bp的平均数据),并计算每个位置相对于该平均读数深度的倍数变化。这意味着侧翼应该从1开始,如果在转录起始位点(基因组的高度开放区域)有高的读数信号,那么信号应该增加,直到中间的一个峰值。我们把这个归一化后的分布中心的信号值作为我们的TSS富集度量。用于评估ATAC-seq。

文库复杂度测量:
理想状态值是: NRF>0.9, PBC1>0.9, and PBC2>3.
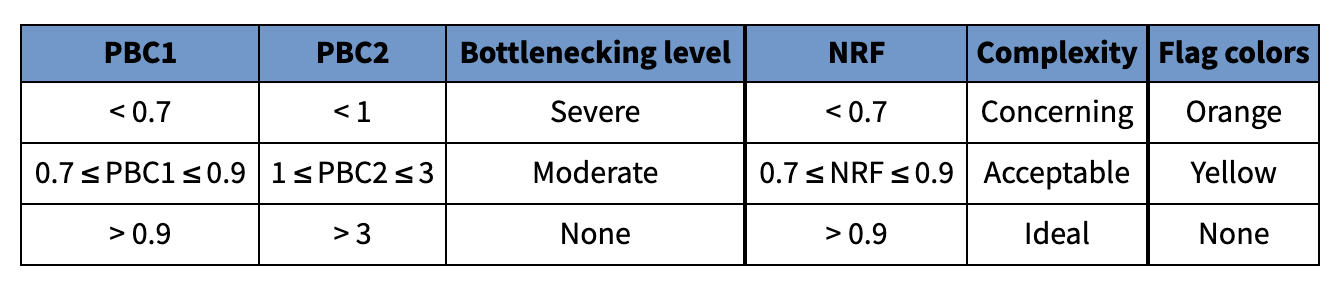
Non-Redundant Fraction (NRF) – Number of distinct uniquely mapping reads (i.e. after removing duplicates) / Total number of reads.
PCR Bottlenecking Coefficient 1 (PBC1)
PCR Bottlenecking Coefficient 2 (PBC2)
1 | import os |
差异Peak分析
差异peak是分析的第一步,也是基础。根据实验的设计,可以比较两个组之间差异的Peak.
以往的几篇文章都推荐使用Diffbind(Differential binding analysis of ChIP-seq peaksets)
目前没有专门为ATAC设计的差异peak 分析工具,不过他们都是计算该区域的counts数据,归一化,对比两个组之间的差异。
另外HOMER, DBChIP,也能实现同样的需求。
利用Diffbind进行差异Peak分析(PCA,MA,heatmap,Volcano,differ Peak):
1 | library(DiffBind) |
Peak 注释
1 |
|
1 | import subprocess |
Motif
peak注释虽然提供了功能解释,但并没有直接解释底层机制。开放的染色质可以通过转录因子影响转录,转录因子通过识别和结合 DNA 上的特定序列(TFBS:TF 结合位点)来促进转录。而事实上转录因子通过与组蛋白或非组蛋白的竞争以及与辅因子的合作来调节转录。
有两种类型的基序或基于 TF 的分析方法(研究TF调控):
- 基序频率或活动的基于序列的预测
- TF 占用的足迹。
JASPAR是现在用的最多的一个motif 数据库,事实上存的就是一些转录因子对应的位置权重矩阵(PWM),其中有的是实验的结果,还有的是计算预测出来的。
工具:
- TFBSTools
- HOMER
- MEME FIMO
原理都是一样的,基于PWM矩阵,然后在序列里面扫描搜索。
一直有一个疑问,对于motif扫描的时候,我们是应该用差异的区域,还是全部的区域?。差异的区域中全部的还是仅上调的区域?
1 | /usr/local/share/bio/homer/bin/findMotifsGenome.pl out.bed hg38 first -len 6,8,10,12,14 |
对于hommer,其result 有两个,一部分是能够和已有数据库中,匹配到的,另外一部分是基于序列预测出来了的,可能没有任何的生物学意义。
对于HINT 也发现能够做motif 富集。
1 | rgt-motifanalysis --enrichment --organism mm9 --input-matrix Matrix_CDP_cDC.txt match/random_regions.bed |
TF Footprints
除了motif,TF Footprints是另外一种研究转录因子调控的方法。原理是TF 与 DNA 结合会阻止结合位点内的 Tn5 切割,就会形成一个深渊低谷一样的峰值分布。
对于检测方法,目前都是基于Boyle 提出的变种隐马尔可夫模型HMM,即在每个碱基使用归一化和平滑的片段计数来检测不同的状态,例如足迹、侧翼和背景。其中目前用的比较多的是针对ATAC数据的HINT-ATAC。
最近的 HINT-ATAC 也使用 HMM,但只有 HINT-ATAC 校正了链特异性 Tn5 切割偏差.
1 | need bam and bed file for input |
@todo 针对单组数据,对于重复数据,两组,还代解决code。
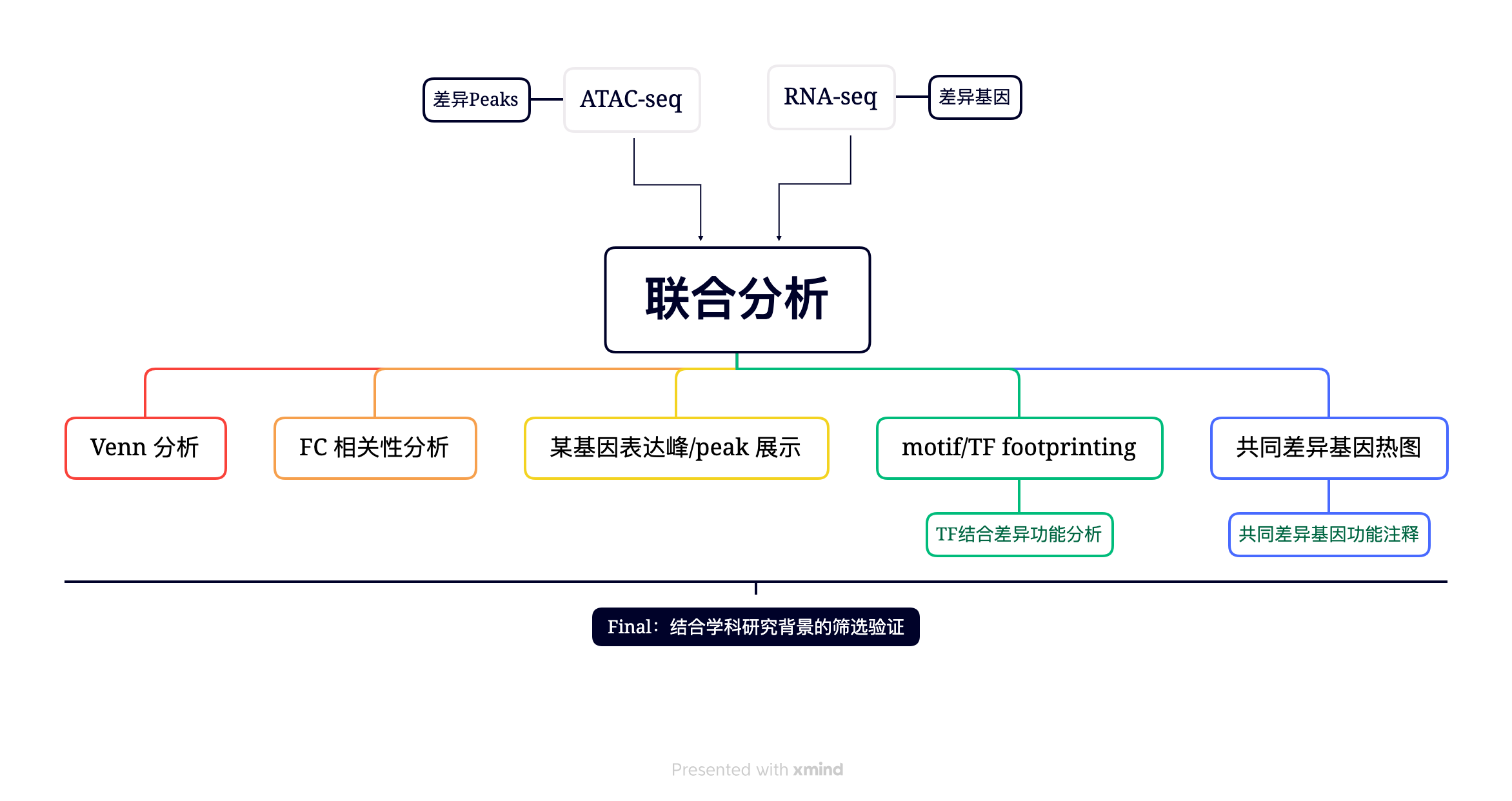
RNA-seq 联合分析
通过 RNA-seq 定性或定量地将染色质可及性的变化与感兴趣的基因表达的变化联系起来,直观地,我们可以发现 DE 基因是否在相应的 TSS 周围也具有显着差异的染色质可及性,可以推断 DE 基因受与开放染色质中特定基序或足迹相关的 TF 调节.
案例:
1. PECA:转录因子TF,染色质调控因子CR和调控元件RE相互作用网络推断的新方法(Cell Stem Cell 2019 )
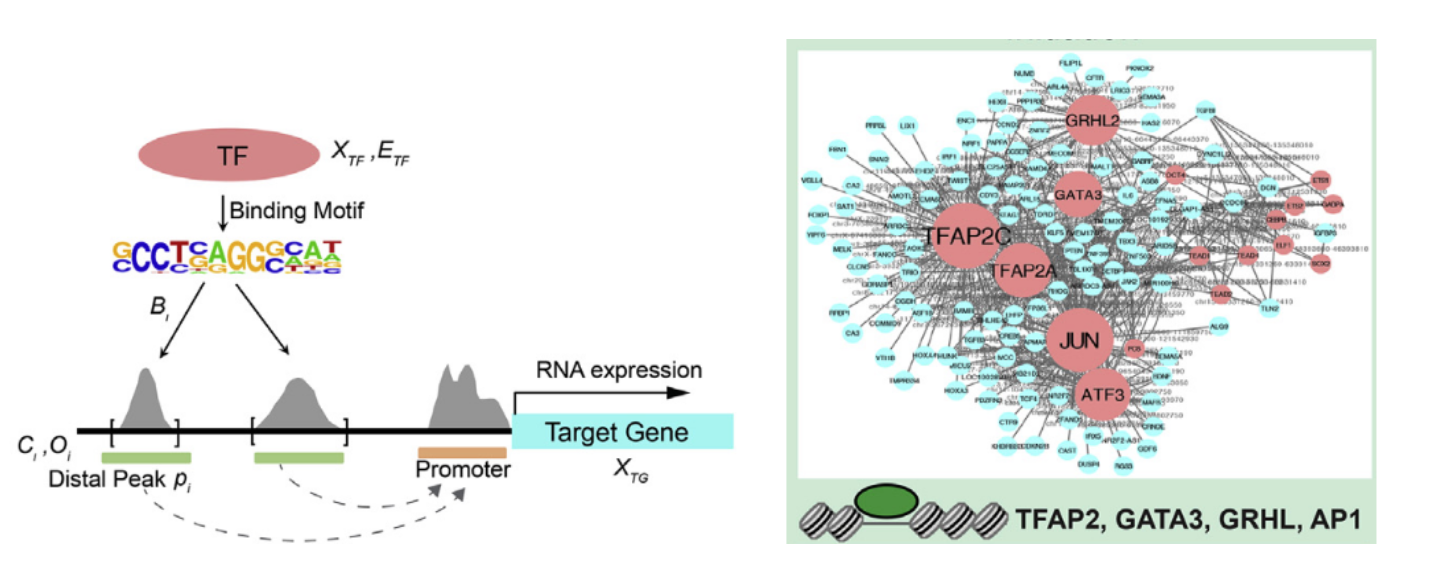
可以使用 PECA[7] 方法重建调控网络。中科院王勇教授团队,利用匹配的基因表达和染色质可及性数据刻画转录因子和调控元件结合调控下游基因表达的数学模型,构建了描绘细胞状态转化的染色质调控网络,通过网络分析鉴定出TFAP2C和p63分别为表面外胚层起始和角质形成细胞成熟的关键因子.
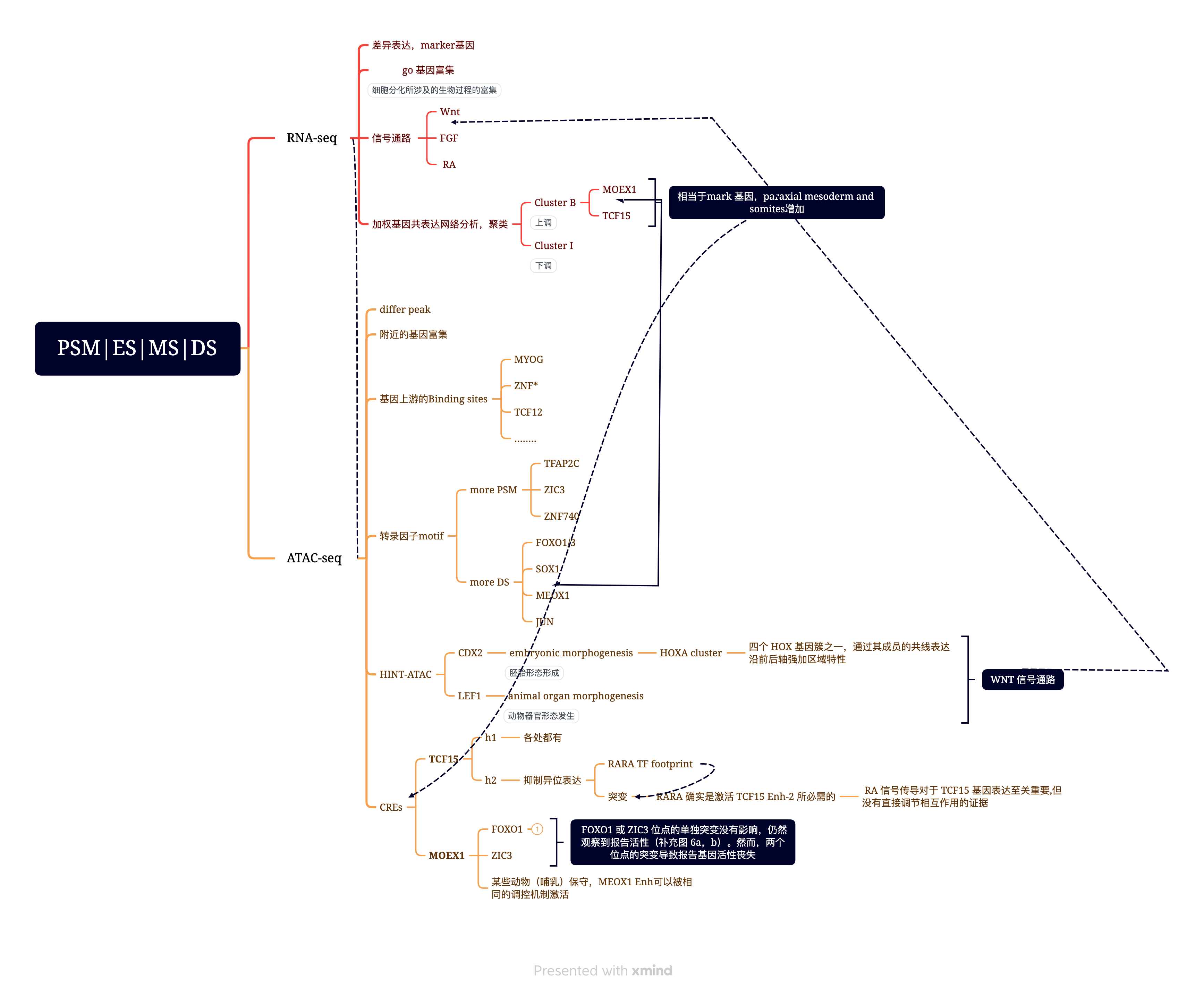
2. 鸡胚的体节分化过程,挖掘关键的TF和Enhancer(nature communications 2021)
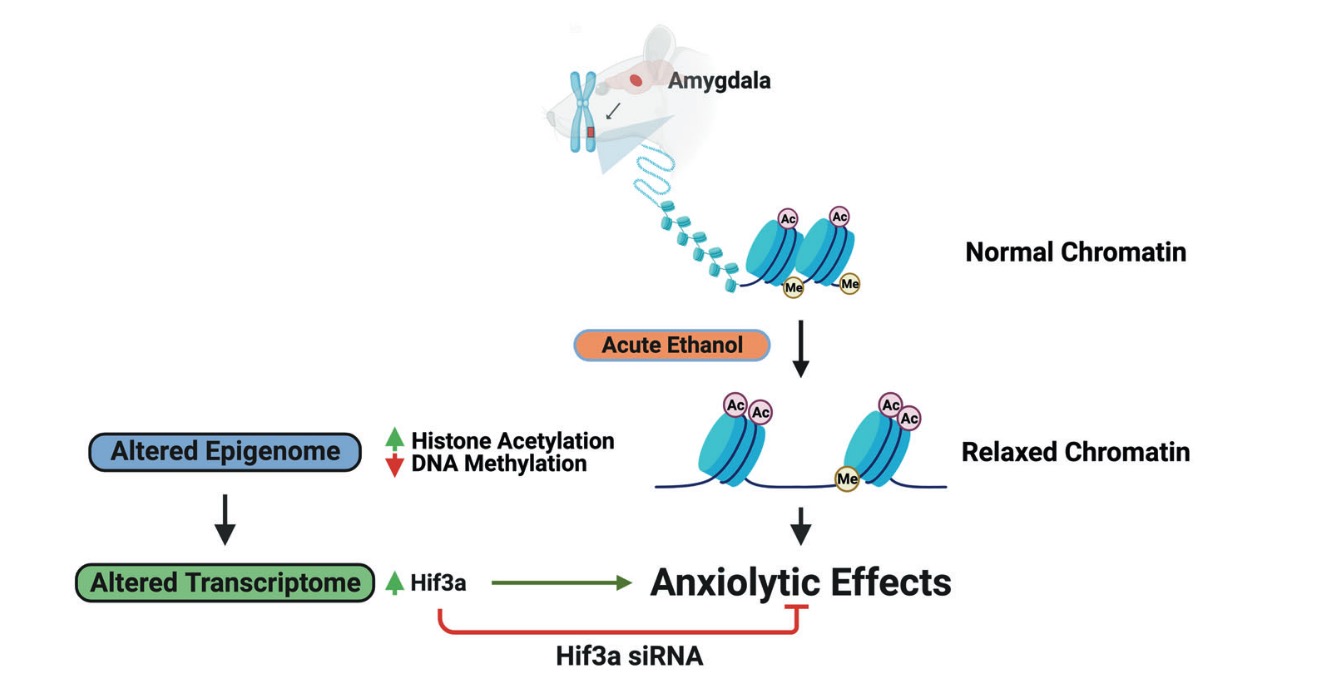
3. 揭示酒精诱导的抗焦虑过程中的表观基因组学和转录组学相互作用(Molecular P s ychiatry 2022)
featureCounts
1 | featureCounts -T 16 -p -t exon -g gene_id -a /home/wvdon/atac/gene/Homo_sapiens.GRCh38.106.gtf -o all_new_feature.txt \ |
RNA-seq 数据分析(差异基因,火山图,热图,富集分析)
1 | base <- read.table("/media/wvdon/sdata/atac-seq/before/all_new_feature.txt",row.names = 1 ,header=T,sep = '\t') |
总结
参考
- Yan, Feng, et al. “From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis.” Genome biology 21.1 (2020): 1-16.
- Krishnan, H. R. et al. Unraveling the epigenomic and transcriptomic interplay during alcohol-induced anxiolysis. Mol. Psychiatry (2022) doi:10.1038/s41380-022-01732-2.
- Mok, G. F. et al. Characterising open chromatin in chick embryos identifies cis-regulatory elements important for paraxial mesoderm formation and axis extension. Nat. Commun. 12, 1157 (2021).
- RS ∗, GB †. DiffBind: Differential binding analysis of ChIP- Seq peak data.
- Li, Z., Schulz, M. H., Look, T., Begemann, M., Zenke, M., & Costa, I. G. (2019). Identification of transcription factor binding sites using ATAC-seq. Genome Biology, 20(1), 45.
- Duren Z, Chen X, Xin J, et al. Time course regulatory analysis based on paired expression and chromatin accessibility data[J]. Genome research, 2020, 30(4): 622-634.
- Li, Lingjie, et al. “TFAP2C-and p63-dependent networks sequentially rearrange chromatin landscapes to drive human epidermal lineage commitment.” Cell Stem Cell 24.2 (2019): 271-284
- Quinlan, AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26:841-842
- “肠化”到底是怎么回事?什么情况下会癌变?https://view.inews.qq.com/a/20210205A0CTTC00
Software Version
| macs2 ==2.2.4 | |
|---|---|
| bwa ==0.7.17 | |
| bowtie2 ==2.3.4.3 | |
| pipeline (v2.1.3) | |
| Homer | |
| HINT-ATAC | |
所有的代码都被上传到GitHub,https://github.com/wvdon/BIO_ATAC
目前是个人私人仓库,后续会开放。